In August 2013 I joined CCP-SAS, a joint US/UK collaborative software development project to provide the infrastructure and tools for analysing small angle scattering data on complex systems using atomistic and coarse grained modeling approaches.

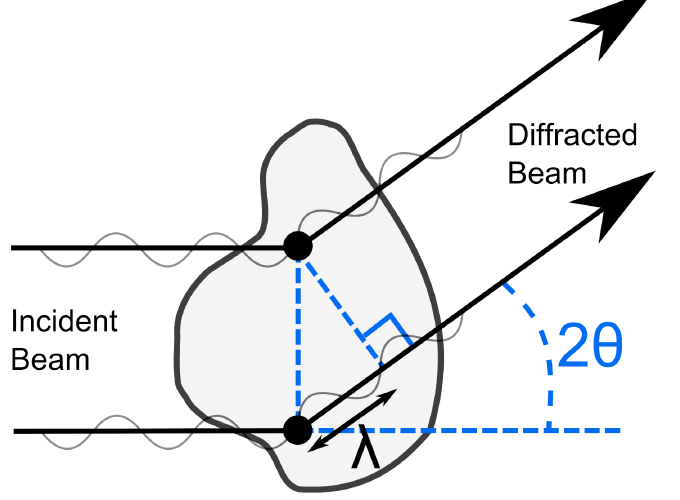
Small angle X-ray and neutron scattering techniques (known as SAXS and SANS respectively, or collectively as small angle scattering, SAS) characterize proteins in solution and complement high-resolution structural studies. They are of particular utility when large proteins cannot be crystallized and in systems where solution conditions affect the structure adopted. Atomistic models of the average structure can be generated through constrained modelling, a technique in which known structures (such as domain or subunit crystal structures) are combined with linker models to produce candidate global conformations. By randomizing the configuration adopted by the different elements of the model thousands of candidate structures can be produced. Theoretical scattering curves are then generated for each model and used for trial-and-error fits to the experimental data. From this a small family of best-fit models can be identified.
Within this project I am not only writing software but also conducting my own research as well as facilitating the use of a variety of modelling techniques (some of which we are developing in CCP-SAS) in other peoples SAS based research. Currently I am investigating monomer IgA1 and developing tools to build antibody structures incorporating carbohydrates. I am also hoping to contribute to the SASSIE tool which is designed to generate and manipulate large numbers of atomistic structures and to calculate the SANS, SAXS, and neutron reflectivity profiles from them.
Update: In the time since I first wrote this post I did infact contribute to SASSIE, in particular on the PDB Scan and PDB Rx modules for evaluating and completeing atomistic structures in perparation for modelling. These tools are available online in SASSIE-web.
SCT: Comparing Atomistic Models to SAS Data
In the Structural Immunology Group at UCL we have developed a suite of tools, called SCT, in order to facilitate both the computation of theoretical scattering curves from atomistic models and their comparison to experiment. The SCT suite also includes programs which add a hydration layer to models, necessary for comparison to data from X-ray scattering, and provide sequence based estimates of protein volume (both incorporating hydration and not).

The original SCT software, written in Fortran, has been used in the production of 71 structures (18 antibodies, 27 complement proteins and 24 oligosaccharides) deposited in the Protein Data Bank between 1998-2013. For the first time we are now making this software publicly available, alongside an easier-to-use reimplementation of the same algorithms in Python. SCT has now been made available under the Apache 2.0 license via GitHub.
- D. W. Wright & S. J. Perkins, “SCT: a suite of programs for comparing atomistic models with small-angle scattering data”, J. Appl. Cryst., 2015, 48, DOI: 10.1107/S1600576715007062
