
The free energy change of biomolecular association (sometimes known as binding affinity) is arguably one of the most important physical quantities in biochemistry, governing the myriad processes that occur in living organisms as well as being one of the pivotal determinants in biomedical therapy and pharmaceutical activity. The use of molecular dynamics simulations to compute accurate estimates of binding affinity is a major field of research. I have been heavily involved in research aiming to use such techniques to evaluate the binding affinity of different variants of HIV-1 protease to drugs designed to inhibit it. The emergence of drug resistance is a major challenge for the effective treatment of HIV and much of my work in this field has been performed in projects (such as CHAIN and ViroLab) which aim to identify and characterize sequences that are resistant to different drugs.
HIV protease is a good test case for free energy calculations as it is relatively small (it consists of a dimer of two 99 amino acid long chains) and its clinical relevance has resulted in it being extensively studied both experimentally and computationally. If rapid and accurate assessments of binding affinities of different drugs to HIV protease could be made these could have direct application in drug discovery and even inform clinical decision support systems.
Ranking of Clinically Relevant Protease Inhibitors
Having previously shown the ability to rank mutant HIV-1 proteases to the single inhibitor lopinavir (see below), we expanded our work to investigate all nine FDA-approved HIV-1 protease inhibitors. As part of this work we also performed a detailed analysis of the performance of MMPBSA and the related molecular mechanics generalized Born surface area (MMGBSA) free energy calculation methods. We confirmed our previous results observing that ensemble simulations allow the computation of converged and reproducible free energy estimates. This comes despite our results showing that the values obtained from replica simulations of the same protease−drug complex, differing only in initially assigned atom velocities, can vary by as much as 10 kcal/mol, which is greater than the difference between the best and worst binding inhibitors under investigation.
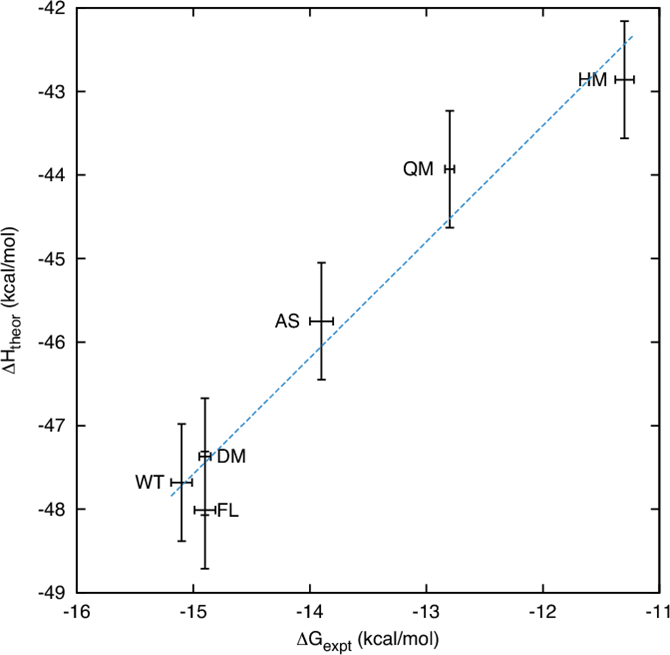
For seven inhibitors, we find that with correctly converged normal mode estimates of the configurational entropy, we can correctly distinguish inhibitors in agreement with experimental data for both the MMPBSA and MMGBSA methods and thus have the ability to rank the efficacy of binding of this selection of drugs to the protease (no account is made for free energy penalties associated with protein distortion leading to the over estimation of the binding strength of the two largest inhibitors ritonavir and atazanavir). We obtain improved rankings and estimates of the relative binding strengths of the drugs by using a novel combination of MMPBSA/MMGBSA with normal mode entropy estimates and the free energy of association calculated directly from simulation trajectories.
- D. W. Wright, B. A. Hall, O. A. Kenway, S. Jha and P. V. Coveney, “Computing Clinically Relevant Binding Free Energies of HIV-1 Protease Inhibitors”, Journal of Chemical Theory and Computation, 2014, 10 (3), DOI:10.1021/ct4007037
One of my co-authors had a protease model 3D printed and helped this work come to the attention of the BBC. One day I will have one of my own, until then I will remain jealous.
Correct Ranking of Lopinavir Binding Strength to Protease Mutants
In order for molecular dynamics simulations to be useful in any real application, the results derived from them need to be well conserved and reproducible. We have found that the necessary sampling required to achieve this goal is most efficiently acquired by using many short simulations rather than single long ones. We used this observation to gain well converged binding free energy estimates for the drug lopinavir bound to sequences of HIV-1 protease, which have been shown experimentally to exhibit a range of binding strengths.
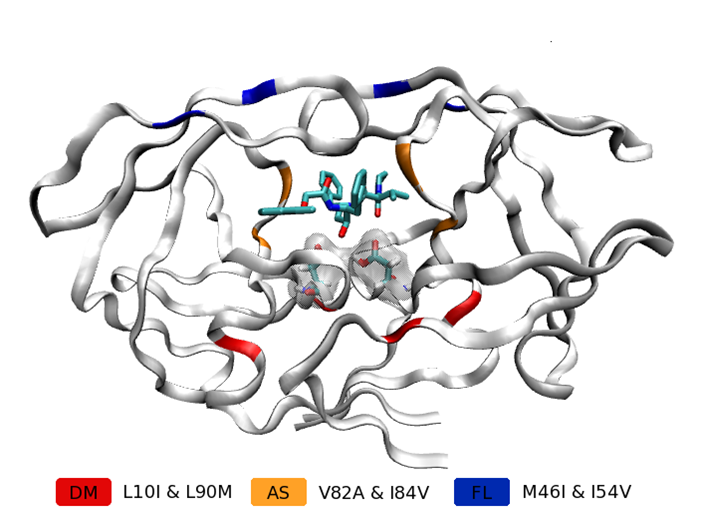
A variety of free energy calculation techniques are available but the requirement for rapid answers in both clinical and drug discovery applications have led us to use the molecular mechanics Poisson−Boltzmann surface area (MMPBSA) method which reduces the cost of calculations at the expense of being less theoretically rigorous than some other methods. Using this approach we produced a ranking of 6 HIV-1 variants to the inhibitor lopinavir with a correlation coefficient of 0.98 (shown to the left, the locations of the mutations in the different variants are shown in the picture above). Incorporating normal mode estimates of conformational entropy allows us to obtain a completely correct rank ordering but with a reduced correlation coefficient of 0.89. To obtain these results, we used ensembles of 50 simulations each run for 6 ns. The changes in binding affinity were seen to correlate strongly with entry of additional water molecules into the protease active site.
- S. K. Sadiq, D. W. Wright, O. A. Kenway and P.V. Coveney, “Accurate Ensemble Molecular Dynamics Binding Free Energy Ranking of Multidrug-Resistant HIV-1 Proteases”, Journal of Chemical Information and Modeling, 2010, 50 (5), DOI: 10.1021/ci100007w
Different Resistance Mechanisms
We have shown that the water entry mechanism of resistance observed in our simulations of lopinavir bound HIV-1 protease also applies in the case of the related inhibitor ritonavir.
- B. A. Hall, D. W. Wright, S. Jha and P. V. Coveney, “Quantized water access to the HIV-1 protease active site as a mechanism for cooperative changes in drug affinity”, Biochemistry, 2012, 51 (33), DOI: 10.1021/bi300432u
However, a study based on a patient derived sequence has shown that we can also use ensemble molecular dynamics simulations to characterize other resistance pathways. In particular, I have shown that mutations at position 71 cause changes in the relative conformation of the two beta sheets that form the protease dimer interface. These changes are associated with changes in drug binding affinity and in some cases enzymatic efficacy.
- D. W. Wright, and P. V. Coveney, “Resolution of Discordant HIV-1 Protease Resistance Rankings Using Molecular Dynamics Simulations”, Journal of Chemical Information and Modeling, 2011, 51 (10), DOI: 10.1021/ci200308r

Automation of Simulation and Analysis
The preparation execution and free energy analysis of the simulations in all the studies described on this page were automated using a set of scripts known as binding affinity calculator (BAC).
- S. K. Sadiq, D. W. Wright, S. J. Watson, S. J. Zasada and P. V. Coveney, “Automated Molecular Simulation Based Binding Affinity Calculator for Ligand-Bound HIV-1 Proteases”, 2008, 48 (9), DOI: 10.1021/ci8000937
We are now working to update this infrastructure to make use of the Simple API for Grid Applications (SAGA) and the extension BigJob which allows the simple distribution of simulations within an ensemble across multiple computers.
